Esophageal atresia
introduction
An esophageal atresia is a congenital malformation (atresia) of the esophagus, which is known in technical terms as the esophagus. This causes a break in the continuity of the esophagus. This interruption of continuity can have different lengths. The length is usually given in centimeters or in the number of vertebral body heights of the children concerned.
When dividing into a short-haul and a long stretched However, there is no real agreement in the literature about esophageal atresia. The morphological classification (morphology = science of the structure and organization of living beings and their components). takes place after Vogt and takes into account the length, the Art the malformation and a possible Fistula formation(Fistula = channel created by disease or artificial that connects an organ with the surface of the body or another organ. The latter is very common, so that 85% of the lower end of the esophagus goes into the windpipe flows out. Typically, esophageal atresia is associated with other congenital malformations.
Read more on the topic Fistulas
causes
The development of an esophageal atresia takes place in the Embryonic period instead of. To understand how this malformation occurs, it is advisable to visualize the natural development of the fetal period. In physiological development, the esophagus is formed from the fetal foregut, which extends from the throat (Pharynx) to stomach extends. From the 20th day of pregnancy, a thickening forms on the anterior edge of this foregut, in which parts of the later trachea begin to differentiate. This part is known as respiratory epithelium. Up to the 26th day of pregnancy, two tubes develop from this structure, namely the esophagus and the trachea, which pass through the Esophagotracheal septum, a kind of partition, can be completely separated from each other. If this separation process interferes with, esophageal atresia can result.
Classification according to Vogt
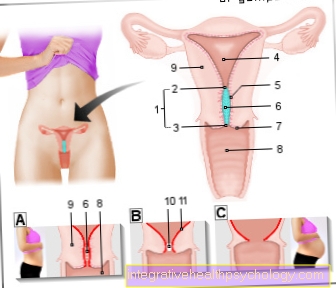
The various forms of esophageal atresia are classified according to Vogt's classification. This division has existed since 1929. One differentiates four types of esophageal atresia. The classification takes into account the presence of a fistula to the windpipe and an atresia (malformation) or Aplasia (complete absence) of the esophagus.
At the Vogt type I. it is an esophageal aplasia. Of the The esophagus is completely absent. At around 1%, this malformation is very rare.
Of the Vogt type II describes a long esophageal atresia without esophagotracheal fistula formation and makes up about 8% of the total.
The Vogt type III is divided into one Type IIIa, b and c. An esophageal atresia with an upper esophagotracheal fistula referred to as type III a. The lower end of the esophagus ends blindly here. With a frequency of> 1%, this type is very rare.
The most common expression says the bailiff Type IIIb represent, which approx 85% of the whole. It is an esophageal atresia with a lower esophagotracheal fistula.
At the Type Vogt IIIc there is an esophagotracheal fistula on both the upper and lower segments. This expression occurs with a frequency of around 5%.
As so-called H-Fistel is called the Vogt Type IV. It is an esophagotracheal fistula without atresia. Their frequency is around 2%.
Occurrence
The esophageal atresia is a congenital malformation that occurs with a frequency of approx 1: 3500 live births goes hand in hand worldwide. At 60%, boys are slightly more affected than girls. The most common of these is the Type III b according to Vogt, namely the esophageal atresia with lower Esophagotracheal fistula formation (So the lower end of the esophagus opens into the trachea). This expression occurs in 85% of the cases.
The remaining forms according to Vogt occur with less than 8% and are rather rare. Familiar accumulation, i.e. the occurrence of the malformation in several family members, is rare. There is a 1% probability of illness in siblings, and 9% in identical twins. The malformation occurs sporadically, so it cannot be assigned to a specific genetic location.
Congenital esophageal atresia is often associated with other congenital malformations. About 50% of children with esophageal atresia also have other malformations. The so-called should be mentioned VACTERL association, the cause of which is largely unknown. It describes a combination of certain malformations. In detail, these are malformations of the Spine (Vertebral), des Anal area (Anal), des Heart (Cor), the air- and esophagus in terms of an esophageal atresia with esophagotracheal fistula formation (trachea and esophagus), the Kidneys (Renal) and finally the limbs (Limb).
diagnosis
Ultrasound may reveal polyhydramnios in the mother before birth. This means that there is an above-average amount of amniotic fluid. However, this marker is relatively unspecific, so that esophageal atresia cannot be proven by this finding alone.
After the birth, attempts are made to insert a nasogastric tube. The feeding tube can only be advanced about 11 to 12 cm until a resilient resistance is felt. Aspiration (material penetrates the trachea) of gastric juice through the gastric tube is also not possible. If air is blown in via the gastric tube, a gurgling sound can be heard with the stethoscope over the shoulder blades and the jugulum (indentation on the upper edge of the breastbone), but not over the stomach. This is where the sound should normally be heard.
To prove the diagnosis, an X-ray examination of the chest and abdomen (chest / abdomen X-ray) is carried out. There are then different changes that result from the different forms of esophageal atresia. An accumulation of air at the level of the 3rd thoracic vertebra in the X-ray image reflects the filling of air in the upper blind sac. If there is a lower fistula, a gas filling can be seen in the stomach and intestines. This is because air escapes through the fistula from the airways into the digestive tract.
Read more on the topic: Chest x-ray (chest x-ray)
In Vogt I, II and IIIa there is no gas filling of the stomach because there is no lower esophagotracheal fistula. A contrast agent examination is rarely carried out in which a contrast agent is carried over if a fistula is present.A contrast agent examination is carried out if the situation and the type of esophageal atresia are not clear from the normal X-ray examination. A water-soluble contrast medium is used for this and the X-ray examination is carried out with it. Further diagnostics include an echocardiography (ultrasound examination of the heart) and an ultrasound examination of the abdomen. On the one hand, this is used for preoperative planning, and on the other hand, further malformations can be found.
Symptoms
There are certain prenatal (before birth) and postnatal (after birth) signs and symptoms that suggest the presence of esophageal atresia.
Before birth, a so-called polyhydramnios, a Above-average amount of amniotic fluid. This is due to the fact that the fetus cannot swallow the amniotic fluid because of the malformation. However, this is a unspecific character and does not necessarily imply the presence of esophageal atresia. The affected babies are mostly premature babies who are noticeable after birth with coughing fits and increased salivation. The saliva runs out of the mouth and collects frothy in front of it. He cannot be swallowed. Choking out the foamy saliva is also typical. The general condition of the babies is also deteriorating. Rattling breathing is common. Furthermore, the children fail, especially when trying to feed cyanosis on. The cyanosis manifests itself by a blue coloration of the skin and mucous membranes, which is caused by the fact that the food does not get into the stomach, but is aspirated into the lungs. This hampers the children's breathing. Feeding should not be attempted if esophageal atresia is suspected. An attempt should be made to insert a feeding tube. However, it is not possible to insert the gastric tube due to the malformation.
Read more on the topic Premature birth
In Vogt type IV esophageal atresia, infants suffer from repeated aspiration pneumonia without showing any further symptoms. Are aspiration pneumonia Pneumoniacaused by repeated inhalation of food particles.
Surgical treatment of esophageal atresia
Surgical therapy is the obligatory measure in the presence of esophageal atresia. The operation is not carried out as an emergency operation, but within the first 48 hours after the birth.
This is an exception Respiratory distress syndrome or one massive overinflation of the stomach with the risk of one Rupture (rupture of an organ) Then an operation is carried out immediately. The primary operation may be delayed if the birth weight is very low or the child is unstable. The primary goal of surgery is to restore a continuous esophagus and to occlude an esophagotracheal fistula, if any. The procedure chosen depends on the type of malformation.
It is usually accessed via a small, right-sided, vertical incision in the right armpit. If there is not a large distance between the malformed sections of the esophagus, the two ends of the Holhorgan can be connected to one another via an end-to-end anastomosis. This means that you connect the two ends with a seam. If the distance between the ends or the length of the esophagus is too short, the organ can pass through a Organ elevation be replaced. For example, the stomach can be pulled up and connected to the rest of the esophagus so that a functional esophagus can be recreated. However, stretching procedures and the natural growth of the esophagus are also used to achieve a sufficient length of it.
To bridge the gap, a so-called salivary fistula is then applied to the newborn until the definitive surgery date. This is one artificial gastric outlet. If there is a fistula formation in the windpipe, it must be surgically cut and closed, otherwise food particles can get into the lungs. That would lead to constant pneumonia and destruction of the lungs. Intensive medical care takes place after the operation. The children are usually ventilated for about 2 to 3 days after the operation. Depending on the course, they are then fed relatively quickly (also after 2 to 3 days) via the gastric tube. After about 10 to 12 days, a contrast agent examination is carried out to assess the success of the operation. If everything goes well, the child will be fed orally.
consequences
The condition following esophageal atresia requires professional follow-up treatment in the first few years. The forecast(Prospect of healing) is true Well, however, there are a number of postoperative complications to be expected.
About 40% of children do gastroesophageal reflux (Stomach acid flows back into the esophagus), which is common in small children bronchopulmonary infections (Infections that affect the lungs and bronchi) favors. The gastric acid reflux promotes the aspiration of small food particles into the windpipe, which leads to the infections. In some cases an operation (Fundoplication) necessary at the stomach entrance. This operation ensures that stomach acid can no longer flow back into the esophagus.
Another consequence is difficult food intake. Especially when switching from liquid to solid food, it can be due to a Anastomotic tightness (30-40%) encounter difficulties. An anastomotic narrowing or anastomotic stenosis is a narrowing and / or closure of the operative connection between the ends of the esophagus. These constrictions are caused by the scarred structure of the operated tissue. If this is the case, an expansion is necessary, which is carried out under general anesthesia. Overall, however, the quality of life of children with an operated esophageal atresia can be described as very good.