Storage diseases
definition
The term storage disease encompasses a number of diseases in which certain substances are deposited in organs or cells due to disturbed processes in the metabolism.
Depending on the substance and organ, storage diseases can vary greatly in their severity and form.
Some storage diseases are already apparent at birth and require immediate therapy, whereas others only appear in the course of life.

What storage diseases are there?
-
Iron storage disease - hemochromatosis
-
Copper Storage Disease - Wilson Disease
-
Protein storage disease
-
Glycogen storage disease
-
Lysosomal storage disease
-
Cholesterol ester storage disease
-
Myocardial storage disease
-
Neutral fat storage disease
Iron storage disease
Iron storage disease, known in specialist circles as hemochromatosis, is a metabolic disorder in which there is an increased deposition of iron in the body and in certain organs.
In most cases, iron storage disease is a hereditary defect that leads to an excessive absorption of iron from the gastrointestinal tract.
The excess iron that is absorbed cannot be excreted as quickly as it is absorbed and is therefore deposited in various organs.
Depending on the affected organ and the amount of iron, a variety of symptoms of hemochromatosis and complaints can occur.
In rare cases, iron storage disease can also occur as a result of another underlying disease or as a result of frequent blood transfusions, known as secondary hemochromatosis.
The additional iron stored in organs, which normally do not serve as iron stores, leads to remodeling processes.
During these remodeling processes, a form of scar tissue is created, which replaces the healthy organ tissue and thus reduces the functionality of the organ.
Frequently, this mainly affects hormone-producing organs in the abdomen, such as the liver (most often) or the pancreas.
Organs such as the heart, skin and pituitary gland are also some of the most frequently damaged organs.
The course of the disease is usually insidious and is therefore often only noticed at an advanced stage of the disease.
The symptoms depend on the extent of the damage and the affected organs.
General symptoms such as fatigue and tiredness are typical at the beginning.
In the course of the disease, there are usually joint pain in the finger joints, especially the index and middle fingers, as well as a noticeable brown coloration of the skin.
With the help of blood tests and special biopsies of individual organs, a precise diagnosis can be made as to which organs are affected and to what extent.
The most common and typical organ manifestations are primarily the liver with liver cirrhosis, which is a high risk factor for the development of liver cancer, and the pancreas with the development of diabetes mellitus.
The options for treating iron storage disease are limited to regular excretion of excess iron.
A causal cure is not yet known.
First and foremost, a low-iron diet is recommended, as well as regular consumption of black tea, as this reduces the absorption of iron in the intestines.
If the iron values are elevated despite a low-iron diet, bloodletting is the method of choice.
Here, 500 ml of blood are taken from the patient, whereby iron bound to the blood cells is lost.
After reaching the target level for iron in the blood, bloodletting every 2-3 months with close laboratory checks is recommended.
This intervention can often be dispensed with in women of childbearing age, since menstrual bleeding leads to sufficient iron loss.
As an alternative to bloodletting, iron-binding drugs are also available, but these are only used if bloodletting is not feasible, for example due to anemia - anemia - or another underlying disease.
With early diagnosis and consistent therapy, those affected with iron storage disease have a normal life expectancy.
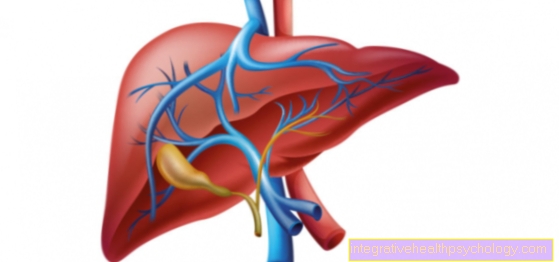
Copper storage disease
The copper storage disease, the so-called Wilson's disease, is a metabolic disease that is based on the disturbed excretion of copper.
The reason for this is a hereditary genetic defect in a protein that prepares copper for excretion in the bile.
If there is a defect here, copper can no longer be excreted in sufficient quantities.
It builds up in the bloodstream and, as a result, is deposited in various organs.
Typically, the copper is mainly deposited in the liver, cornea, red blood cells and the brain.
In particular, the involvement of the liver and the brain lead in combination to typical symptoms, which lead to the suspected diagnosis of a copper storage disease.
The first symptoms often appear between the ages of 5 and 10, for example in the form of an inflammation of the liver, known as hepatitis, or neurological restrictions due to decreased liver function, such as drowsiness and shaky hands.
From the age of 10, neurological complaints typically appear, such as fine hand tremors, dementia, swallowing or speech disorders, and gait disorders.
In addition, the copper deposits can become visible in the eye.
Here is a green-brown ring in the cornea.
The diagnosis of copper storage disease can be confirmed with the help of blood and urine tests, possibly a biopsy of the liver and various imaging tests.
If the diagnosis of copper storage disease is confirmed, the primary therapy consists of a combination of a low-copper diet and drugs that serve to excrete copper, the so-called chelating agents, e.g. D-penicillamine.
If therapy is started early and consistently, the prognosis for the disease is good.
The only important thing is to make an early diagnosis, before organ damage can occur due to the copper deposits.
The following applies: any unclear liver disease that is not due to an infection, in combination with unclear movement disorders before the age of 45 should be clarified with regard to a copper storage disease.
Also read the article: The genetic test
Protein storage disease
The so-called protein storage disease is not a recognized clinical picture according to the World Health Organization.
Rather, it is a concept that Prof. Dr. Lothar Wendt was developed and published.
In his work, Prof. Wendt pursued an alternative approach to explaining common diseases in our society, contrasting the view of traditional medicine with the question of “why”.
A typical example of this approach can be illustrated by the common disease diabetes.
In type 2 diabetes mellitus, the blood sugar level is very high.
These increased blood sugar levels lead to damage throughout the body with serious complications.
The conventional medical approach is therefore to consistently lower the blood sugar level in order to prevent further damage.
Prof. Wendt, on the other hand, asks in his working concept why these increased blood sugar levels occur and whether the reason for this could be compensation.
Here he puts forward the theory that protein deposits in the walls of the blood vessels cause them to thicken.
Prof. Wendt explains that the increased blood sugar level is a reaction to the thickened blood vessel walls in order to transport a sufficient amount of sugar into the cell despite the increased resistance and the longer diffusion path.
According to Wendt, it is not the sugar that is the disease-causing factor, but the protein and ultimately the term diabetes is misleading.
The term increased blood sugar level as a result of a causal protein storage disease would be more appropriate according to his concept.
At present, however, there is a lack of evidence-based studies that would support this explanatory approach and the concept of the disease.
Only in the treatment of osteoarthritis are there already those affected in some self-help groups who report that they have alleviated or even eliminated osteoarthritis through targeted protein degradation therapy.
It must be noted, however, that these are individual empirical values without a reference group, which were only successful if the therapy was started at a very early stage of the disease.
Leading professors from various departments see, taking into account the current study situation, no evidence for the correctness of the concept of protein storage disease by Prof. Wendt.
Glycogen storage disease
In glycogen storage diseases, an hereditary genetic defect leads to excessive deposition of glycogen in the body.
Glycogen is also known colloquially as liver starch.
This is a long and multiply branched glucose molecule, which is stored in the liver in particular and serves as a supplier of the energy carrier sugar.
There are a total of nine different forms of glycogen storage disease, each of which is based on a different genetic defect and leads to the deposition of glycogen in different organs.
The most common forms include Glycogen Storage Disease Type I von Gierke Disease, Glycogen Storage Disease Type II, Pompe Disease, and Glycogen Storage Disease Type V, McArdle Disease.
The various forms differ both in their symptoms and in the onset of the disease.
The type I glycogen storage disease is usually noticeable by an enlarged liver and a distended abdomen, in addition there are often seizures and a tendency to bleed.
In type II glycogen storage disease, muscle wasting all over the body and an oversized tongue are particularly noticeable.
In type V glycogen storage disease, too, generalized muscle wasting occurs, but in combination with muscle pain and cramps after exertion.
The therapy of glycogen storage diseases depends on the type of disease and its severity.
Lysosomal storage disease
The term lysosomal storage disease encompasses a large group of diseases which are based on a genetic defect in the lysosomes.
Lysosomes are a group of cells in the human body that act like the stomach or the garbage can of cells.
All excess cell components and waste products of the cell are broken down in the lysosomes.
If the lysosomes are defective, these cell waste products accumulate, which are then deposited both in the cell and in other organs.
45 diseases are currently assigned to the group of lysosomal storage diseases.
Most diseases are very rare variants of the storage disease.
The most common forms of lysosomal storage disease are Gaucher's disease and Fabry's disease.
Read more on the topic: Fabry disease
In Gaucher's disease, the disrupted breakdown processes lead to an accumulation of fats in cells and other organs.
Symptoms vary widely due to the potential for affecting the entire body.
Enlargement of the liver and spleen, disorders in the blood-forming system and seizures are typical.
The disease is often noticeable in infancy due to a feeding disorder.
Fabry disease, on the other hand, is significantly rarer than Gaucher disease and mainly affects boys due to its inheritance.
The symptoms of Fabry's disease initially include burning attacks of pain in the fingers, gastrointestinal complaints and corneal opacity.
Later, the heart can become infected with heart failure and strokes.
Find out more about the topic here: Gaucher's disease.
Cholesterol ester storage disease
Cholesterol ester storage disease belongs to the group of lysosomal storage diseases, which is a rare hereditary metabolic disease.
Cholesterol ester disease is based on a defect in the lysosomal acidic lipase, which normally breaks down fats such as cholesterol esters and triacylglycerides.
The reduced breakdown of these fats leads to an accumulation of fats in the cell and consequently also in the body's circulation.
This disease does not cause any complaints for a long time, only the reactive enlargement of the liver can lead to a feeling of pressure in the right upper abdomen, nausea or a feeling of fullness.
Blood tests show increased blood values for cholesterol and lipids, as well as decreased values for good fats (HDL).
Fatty liver may appear in the ultrasound examination of the upper abdomen.
The treatment of cholesterol ester storage disease takes place with drugs by inhibiting cholesterol uptake with colestyramine or ezetimibe and additionally by lowering blood lipid values with statins such as simvastatin.
Myocardial storage disease
In myocardial storage disease, degradation products are deposited in the heart walls, which can severely restrict the heart's performance and pumping function.
Two different storage diseases can lead to these deposits in the heart walls: the rare and hereditary lysosomal storage disease Fabry disease and the so-called amyloidosis.
In Fabry's disease, an inherited genetic defect leads to a reduced breakdown of metabolic products, which as a result are deposited, among other things, in the heart walls and can lead to severe damage.
Amyloidosis, on the other hand, can be hereditary as well as acquired in the course of life.
With this clinical picture, too, there are deposits of abnormally changed metabolic products, which, in addition to other organs, mainly accumulate in the heart and here severely limit the function of the heart.
A myocardial storage disease becomes noticeable at the beginning by general symptoms such as weakness and fatigue.
Over time, there is an increasing shortage of breath after exercise and at some point also at rest.
Water in the lungs, in the abdomen, in the legs and in the pericardium are typical side effects as the disease progresses.
Imaging procedures and a heart muscle biopsy are necessary for a clear diagnosis of myocardial storage disease.
The subsequent therapy is then based on the underlying disease that caused it.
Neutral fat storage disease
Neutral fat storage diseases are very rare diseases in which the breakdown and storage of a fat, the so-called triglyceride, is defective.
To date, only 50 cases of neutral fat storage diseases have been described worldwide.
As with most storage diseases, the cause of the genetic defect is also hereditary in neutral fat storage disease.
The disease is often noticed in early childhood due to a developmental disorder.
A majority of those affected develop an enlarged liver with an associated liver dysfunction, as well as eye problems and hearing loss.
Muscle wasting and gait disorders can occur in advanced age.

















.jpg)